Painful Papules on the Arms






The Diagnosis: Piloleiomyoma
Leiomyoma cutis, also known as cutaneous leiomyoma, is a benign smooth muscle tumor first described in 1854.1 Cutaneous leiomyoma is comprised of 3 distinct types that depend on the origin of smooth muscle tumor: piloleiomyoma (arrector pili muscle), angioleiomyoma (tunica media of arteries/veins), and genital leiomyoma (dartos muscle of the scrotum and labia majora, erectile muscle of nipple).2 It affects both sexes equally, though some reports have noted an increased prevalence in females. Piloleiomyomas commonly present on the extensor surfaces of the extremities (solitary) and trunk (multiple).1 Tumors most often present as firm flesh-colored or pink-brown papulonodules. They can be linear, dermatomal, segmental, or diffuse, and often are painful. Clinical differential diagnosis for painful skin tumors is aided by the acronym "BLEND AN EGG": blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor.3 For isolated lesions, surgical excision is the treatment of choice. For numerous lesions in which excision would not be feasible, intralesional corticosteroids, medications (eg, calcium channel blockers, alpha blockers, nitroglycerin), and botulinum toxin have been used for pain relief.4
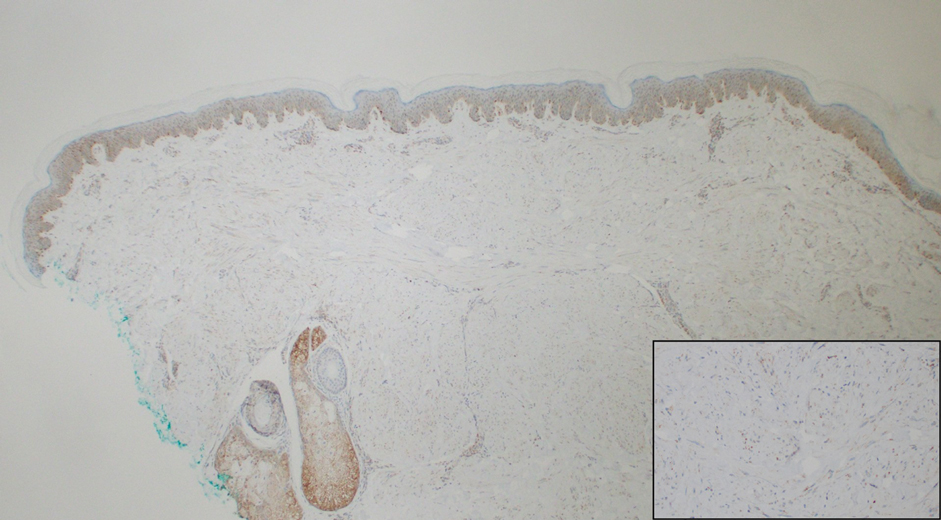
Notably, multiple cutaneous leiomyomas can be seen in association with uterine leiomyomas in Reed syndrome due to an autosomal-dominant or de novo mutation in the fumarate hydratase gene, FH. Reed syndrome is associated with a lifetime risk for renal cell carcinoma (hereditary leiomyomatosis and renal cell cancer) in 15% of cases with FH mutations.5 In our patient, both immunohistochemical staining and blood testing for FH were performed. Immunohistochemistry revealed notably diminished staining with only weak patchy granular cytoplasmic staining present (Figure 1). Genetic testing revealed heterozygosity for a pathogenic variant of the FH gene, consistent with a diagnosis of Reed syndrome.
Histologically, the differential diagnosis includes other spindle cell tumors, such as dermatofibroma, neurofibroma, and dermatomyofibroma. The histologic appearance varies depending on the type, with piloleiomyoma typically located within the reticular dermis with possible subcutaneous extension. Fascicles of eosinophilic smooth muscle cells in an interlacing arrangement often ramify between neighboring dermal collagen; these smooth muscle cells contain cigar-shaped, blunt-ended nuclei with a perinuclear clear vacuole. Marked epidermal hyperplasia is possible.6 A close association with a nearby hair follicle frequently is noted. Although differentiated smooth muscle cells usually are evident on hematoxylin and eosin, positive staining for smooth muscle actin (SMA) and desmin can aid in diagnosis.7 Immunohistochemical staining for FH has proven to be highly specific (97.6%) with moderate sensitivity (70.0%).8 Angioleiomyomas appear as well-demarcated dermal to subcutaneous tumors composed of smooth muscle cells surrounding thick-walled vaculature.9 Scrotal and vulvar leiomyomas are composed of eosinophilic spindle cells, though vulvar leiomyomas have shown epithelioid differentiation.10 Nipple leiomyomas appear similar to piloleiomyomas on histology with interlacing smooth muscle fiber bundles.
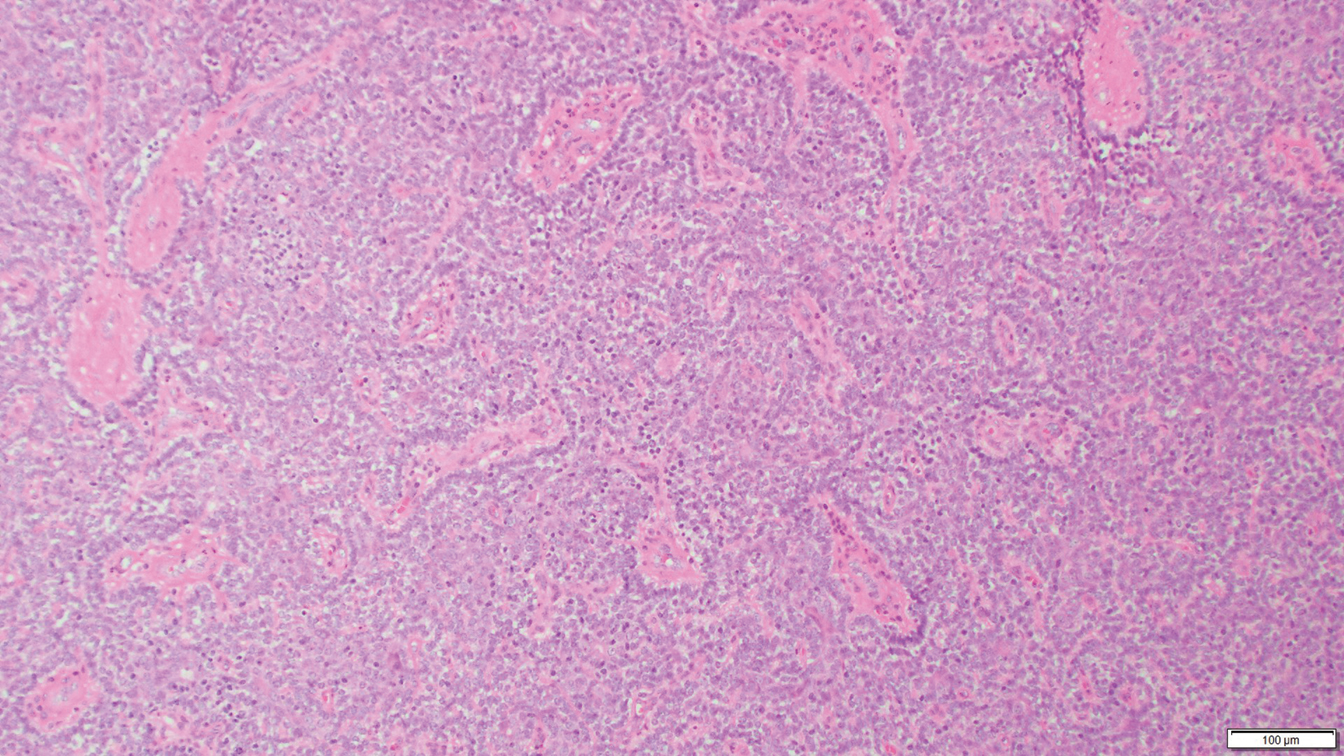
Eccrine spiradenoma is a relatively uncommon adnexal tumor derived from eccrine sweat glands. It most often presents as a small, painful or tender, intradermal nodule (or rarely as nodules) on the head or ventral trunk.11 There is no sexual predilection. It affects adults at any age but most often from 15 to 35 years. Although rare, malignant transformation is possible. Histologically, eccrine spiradenomas appear as a well-demarcated dermal tumor composed of bland basaloid cells with minimal cytoplasm, often with numerous admixed lymphocytes and variably prominent vasculature (Figure 2). Eosinophilic basement membrane material can be seen within or surrounding the nodules of tumor cells. Multiple spiradenomas can occur in the setting of Brooke-Spiegler syndrome, which is an autosomal-dominant disorder due to an inherited mutation in the CYLD gene. Spiradenomas are benign neoplasms, and surgical excision with clear margins is the treatment of choice.12
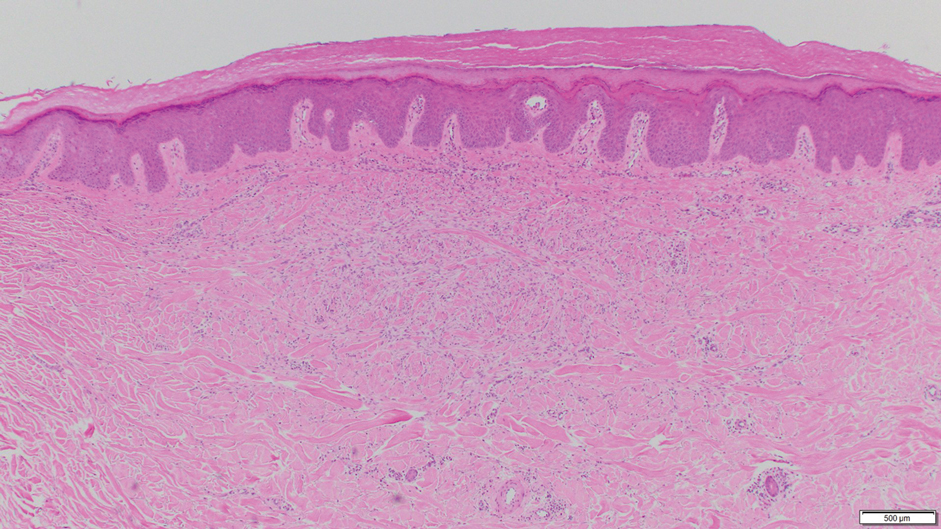
Dermatofibroma, also known as cutaneous benign fibrous histiocytoma, is a firm, flesh-colored papule or nodule that most often presents on the lower extremities. It typically is seen in women aged 20 to 40 years.13 The etiology is uncertain, and dermatofibromas often spontaneously develop, though there are inconsistent reports of development with local trauma including insect bites and puncture wounds. The dimple sign refers to skin dimpling with lateral pressure.13 Most commonly, dermatofibromas consist of a dermal proliferation of bland fibroblastic cells with entrapment of dermal collagen bundles at the periphery of the tumors (Figure 3). The fibroblastic cells often are paler and less eosinophilic than smooth muscle cells seen in cutaneous leiomyomas, with tapered nuclei that lack a perinuclear vacuole. Admixed histocytes and other inflammatory cells often are present. Overlying epidermal hyperplasia and/or hyperpigmentation also may be present. Numerous histologic variants have been described, including cellular, epithelioid, aneurysmal, atypical, and hemosiderotic types.14 Immunohistochemical stains may show patchy positive staining for SMA, but h-caldesmon and desmin typically are negative.
Neurofibroma is a tumor derived from neuromesenchymal tissue with nerve axons. They form through neuromesenchyme (eg, Schwann cells, mast cells, perineural cells, endoneural fibroblast) proliferation. Solitary neurofibromas occur most commonly in adults and have no gender predilection. The most common presentation is an asymptomatic, solitary, soft, flesh-colored papulonodule.15 Clinical variants include pigmented, diffuse, and plexiform, with plexiform neurofibromas almost always being consistent with a diagnosis of neurofibromatosis type 1. Histologically, neurofibromas present as dermal or subcutaneous nodules composed of randomly arranged spindle cells with wavy tapered nuclei within a loose collagenous stroma (Figure 4).16 The spindle cells in neurofibromas will stain positively for S-100 protein and SOX-10 and negatively for SMA and desmin.

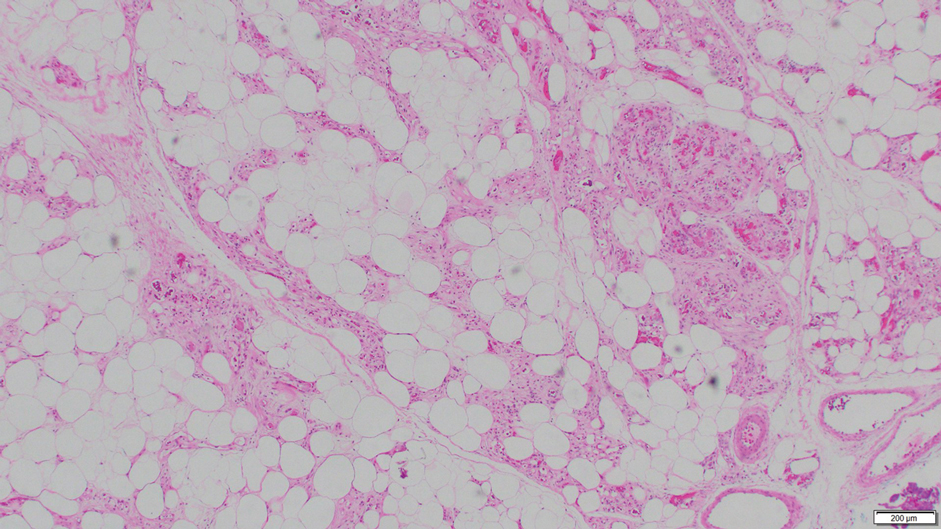
Angiolipoma is a benign tumor composed of adipocytes that also contains vasculature.17 The majority of cases are of unknown etiology, though familial cases have been described. They typically present as multiple painful or tender (differentiating from lipomas) subcutaneous swellings over the forearms in individuals aged 20 to 30 years.18 On histopathology, angiolipomas appear as well-circumscribed subcutaneous tumors containing mature adipocytes intermixed with small capillary vessels, some of which contain luminal fibrin thrombi (Figure 5).