Les coefficients calculés par ce modèle permettent de représenter des azéotropes et des démixtions de phases. Ces deux comportements ne sont pas prédictibles par le modèle de la solution idéale, en particulier par la loi de Raoult. Il s'agit toutefois d'un modèle empirique, dont les paramètres doivent être établis expérimentalement et ne dépendent ni de la température ni de la pression. Ce modèle n'est donc pas prédictif. En pratique, son domaine d'application est limité aux mélanges dont le comportement est proche de celui de la solution idéale.
Dans cette relation, il est supposé que le gaz est un mélange de gaz parfaits et que la variation de la pression totale avec une variation de composition est négligeable. Si les pressions partielles sont écrites sous la forme :
avec et deux fonctions arbitraires ne dépendant que de la température, alors on a :
Dans son article, Margules propose de compléter cette solution en introduisant deux fonctions dépendant de la composition[3] :
Les paramètres et sont liés par les relations :
Ce qui donne :
Margules montre ensuite que cette solution permet de représenter des azéotropes et des démixtions liquide-liquide, ce que ne permettent pas les lois idéales[1].
On considère deux corps purs et dans la même phase liquide ou solide, d'enthalpies libresmolaires respectives et . On mélange une quantité du corps avec une quantité du corps . L'opération étant réalisée à pression et température constantes, le deuxième principe de la thermodynamique implique que l'enthalpie libre ne peut que décroître durant l'opération, c'est-à-dire, pour l'enthalpie libre molaire du mélange résultant, dans la même phase que les corps purs :
d'où, en divisant par la quantité de matière totale , avec les fractions molaires et :
On définit l'enthalpie libre de mélange molaire par la variation d'enthalpie libre molaire due à l'opération de mélange :
Enthalpie libre de mélange molaire :
L'enthalpie libre de mélange molaire ne peut donc être que négative ; elle est nulle pour les corps purs ( pour = 1 et pour = 1). Il est impossible, à pression et température constantes, de réaliser un mélange dans lequel est positive.
Dans un mélange idéal, par définition, il n'y a aucune interaction entre les molécules. La création d'enthalpie libre pour un mélange idéal, ou enthalpie libre de mélange idéale molaire , est donnée par :
Enthalpie libre de mélange idéale molaire :
Cette fonction est toujours négative ou nulle. Le mélange des deux composants est donc toujours possible selon le modèle idéal.
Dans un mélange réel, les interactions entre molécules doivent être prises en compte : la répulsion entre molécules et molécules a tendance à empêcher le mélange, alors que l'attraction entre ces molécules a tendance à le favoriser. Il résulte de ces interactions une enthalpie libre d'excès molaire par rapport au mélange idéal. L'enthalpie libre de mélange molaire réelle vaut ainsi :
Enthalpie libre d'excès molaire :
Pour les corps purs et dans un mélange idéal, par définition. Le modèle d'enthalpie libre d'excès molaire issu des travaux de Margules est de la forme[5],[6] :
des paramètres qui décrivent l'interaction entre et dans le milieu ; sous cette forme, ces paramètres sont adimensionnels.
Le modèle peut également être présenté sous la forme[2],[7],[8] :
Dans ce cas, les paramètres ont la dimension d'une énergiemolaire (en joules par mole)[9]. Cette forme n'est pas retenue dans le reste de l'article.
En toute rigueur, quelle que soit la forme adoptée, les paramètres , , etc. sont des fonctions de la pression et de la température, mais ils sont le plus souvent considérés comme des constantes[6].
Diverses méthodes existent pour généraliser le modèle aux mélanges à trois corps et plus[2],[7]. La forme suivante est souvent utilisée :
généralisation :
Le paramètre introduit les interactions ternaires entre molécules , et , toutes différentes. La méthode de généralisation la plus simple est de négliger ces interactions[5],[6] :
généralisation :
avec le modèle du mélange binaire appliqué aux deux corps et .
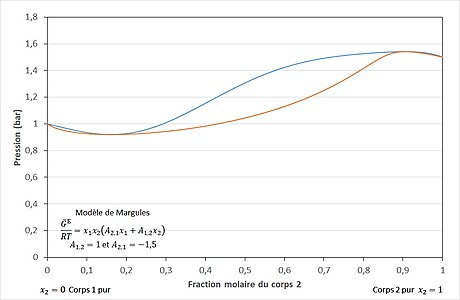
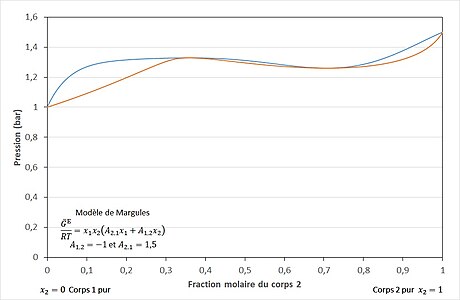
Le modèle de Margules à deux paramètres est un modèle très simple. Il peut être appliqué à de nombreux mélanges. Il peut représenter des équilibres liquide-vapeur ayant une déviation positive (cf. figure 3) comme ceux ayant une déviation négative (cf. figure 6) par rapport au modèle idéal de la loi de Raoult (cf. figure 2). Il représente également les mélanges dans lesquels l'un des coefficients d'activité est plus grand que 1 et l'autre plus petit, et ceux pour lesquels les coefficients atteignent des extrémums[6]. Il peut représenter des azéotropes (cf. figure 4, figure 5, figure 7 et figure 8), des démixtions de phases[10] et des équilibres liquide-liquide-vapeur (cf. figure 15)[11]. Il est largement utilisé en métallurgie pour représenter des phases solides[11].
Le modèle de Margules est cependant empirique. Ses paramètres n'ont aucun sens physique et ne découlent d'aucune théorie. De plus, ce modèle ne dépend pas de la température. Les paramètres ne peuvent donc être établis qu'en régressant des résultats expérimentaux et ne peuvent être utilisés qu'à la température à laquelle ces résultats ont été obtenus. Ce modèle n'est donc pas prédictif.
Ce modèle simple est également incapable de représenter correctement des mélanges fortement non idéaux, dans lesquels les molécules des diverses espèces chimiques ont de fortes interactions[6],[11]. Il ne peut par exemple représenter correctement les liaisons hydrogène, les dimérisations d'acides, les associations entre alcools et dans les solutions aqueuses[6], les démixtions liquide-liquide des mélanges alcools-hydrocarbures[5]. Ses applications se limitent aux solutions relativement simples et aux liquides non polaires[11].
La relation de Gibbs-Duhem induit que les dérivées des deux coefficients d'activité et sont de signes opposés. Les deux coefficients varient donc en sens inverses : si l'un croît, l'autre décroît[12]. La relation induit également que les deux dérivées s'annulent en même temps : les deux coefficients atteignent donc d'éventuels extrémums aux mêmes compositions du liquide. Ces extrémums sont inverses : si l'un des coefficients atteint un maximum, l'autre atteint un minimum. Pour le modèle de Margules à deux paramètres, la dérivée de s'annule en[13] :
Si , alors n'est pas comprise entre 0 et 1 :
si , alors est monotone strictement croissant en (décroissant en ) ;
si , alors est monotone strictement décroissant en (croissant en ).
Si cette dérivée est positive, l'extrémum est un minimum, si elle est négative, c'est un maximum. Si , alors :
si , alors présente un maximum ;
si , alors présente un minimum.
Si , alors :
si , alors présente un minimum ;
si , alors présente un maximum.
Exemple
Le système binaire chloroforme (1)-méthanol (2) montre un maximum du coefficient d'activité du chloroforme à = 0,17, avec les paramètres = 0,6298 et = 1,9522 à 20 °C.
Le modèle peut être simplifié en supposant que , ce qui donne[7] :
Une solution suivant ce modèle est appelée solution régulière[14],[15]. Lorsque = 0 la solution est idéale.
Cette simplification, appelée modèle de Porter[6],[5], rend le modèle symétrique par rapport à la composition = 0,5, ce qui n'est pas réaliste[16]. Elle interdit également la possibilité de représenter des extrémums des coefficients d'activité. Elle est cependant souvent utilisée à des fins pédagogiques pour sa simplicité. Elle est également utilisée dans certaines extensions de la loi de Henry : équation de Krichevsky-Ilinskaya[17],[18] et extension aux mélanges multicomposants[19].
Soit un mélange de deux corps notés et , à température donnée et pression modérée (moins de 10 bar). La phase vapeur est considérée comme un mélange de gaz parfaits, la phase liquide n'est pas idéale. L'équilibre liquide-vapeur est décrit par les équations :
et les fractions molaires des deux corps en phase gaz ().
Dans un mélange azéotropique, la phase gaz a la même composition que la phase liquide :
On obtient pour la pression totale d'un azéotrope[20] :
Il existe donc un azéotrope s'il existe une composition de la phase liquide vérifiant[11],[21],[22] :
Condition générale d'azéotropie :
Cette condition ne peut être remplie par le modèle de la solution idéale (loi de Raoult), dans lequel = 1 (cf. figure 2). Le modèle idéal est donc incapable de représenter un azéotrope.
Pour la plupart des mélanges réels, la pression d'équilibre liquide-vapeur est supérieure à la pression donnée par la loi de Raoult (cf. figure 3). Dans ces conditions, un éventuel azéotrope est dit azéotrope à déviation positive ou azéotrope positif (cf. figure 4). Pour certains mélanges réels, peu fréquents, la pression d'équilibre liquide-vapeur est inférieure à la pression donnée par la loi de Raoult (cf. figure 6). Dans ces conditions, un éventuel azéotrope est dit azéotrope à déviation négative ou azéotrope négatif (cf. figure 7)[21]. Il existe des cas encore plus rares de mélanges présentant deux azéotropes en fonction de la composition, comme le système benzène-hexafluorobenzène[23] (cf. figure 5 et figure 8).
Selon les valeurs des paramètres, cette fonction du second degré en peut avoir jusqu'à deux racines réelles situées entre 0 et 1. Ce modèle peut donc représenter des cas à un ou deux azéotropes. Si la fonction n'a pas de racine réelle, ou si ses racines réelles se situent hors de la plage de composition de 0 à 1, le mélange ne présente pas d'azéotrope, le mélange est dit zéotrope[24]. La figure 1 donne les domaines de zéotropie, de simple et de double azéotropie en fonction des paramètres de Margules[22]. La courbe elliptique qui sépare le domaine de zéotropie de celui de double azéotropie a pour équation[22] :
Le tableau suivant donne des conditions nécessaires (mais pas suffisantes) d'apparition des azéotropes pour le modèle de Margules à deux paramètres[22],[21].
Conditions d'apparition des azéotropes pour le modèle de Margules à deux paramètres.
Cette fonction du premier degré ne peut avoir qu'une seule solution et ne peut donc représenter des cas de double azéotrope. Avec par convention , ce modèle donne[22],[25] :
Figure 9 - Enthalpie libre de mélange molaire avec le modèle d'enthalpie libre d'excès molaire de Margules à un paramètre (solution régulière). Sous la courbe critique, le mélange ne présente qu'une seule phase. Au-dessus de la courbe critique, le mélange présente une démixtion de deux phases[26].
Figure 10 - Quelques courbes d'enthalpie libre de mélange molaire du modèle à deux paramètres de Margules. Pour = 2, les courbes avec = -0,6469 et = 2 sont des courbes de démixtion critiques présentant un point critique.
Par construction, cette condition s'applique aussi à . Ainsi un mélange stable ne peut-il avoir une enthalpie libre de mélange concave. Dans un tel cas, le mélange est instable et se sépare en deux phases ou plus en équilibre, l'enthalpie libre de mélange globale de l'ensemble des phases résultantes étant convexe. Une fonction montrant une démixtion est convexe proche des deux corps purs et concave sur une plage de composition intermédiaire : elle comporte deux changements de convexité, soit deux points d'inflexion. On étudie la fonction enthalpie libre de mélange molaire avec le modèle de Margules à deux paramètres :
La figure 9 ci-dessus montre le tracé de avec le modèle à un paramètre, . Pour = 0 le mélange est idéal. Plus s'éloigne de 0, plus le comportement du mélange s'éloigne du comportement idéal. Pour < 2 la fonction est strictement convexe ; le mélange ne présente qu'une seule phase. Pour > 2 la fonction présente deux points d'inflexion entre lesquels elle est concave ; le mélange présente une démixtion de deux phases[26]. La courbe obtenue avec = 2 est la courbe critique de démixtion ; sur cette courbe, les deux points d'inflexion sont confondus en un point unique appelé point critique de démixtion[26]. Ce modèle ne génère qu'un seul point critique avec = 0,5 et = 2[26]. Le modèle à deux paramètres présente les mêmes types de comportement selon les valeurs des paramètres (cf. figure 10), aux différences près que la fonction n'est pas symétrique par rapport à = 0,5 et qu'il peut représenter une infinité de points critiques (cf. figure 11 et figure 12).
Si la fonction est tracée en fonction de la fraction molaire , le domaine de concavité de la fonction est encadré à gauche par un point d'inflexion où la fonction convexe devient concave, soit , et à droite par un point d'inflexion où la fonction concave redevient convexe, soit . Au point critique de démixtion, les deux dérivées doivent être égales, ce qui impose[10] :
deuxième condition de criticité :
soit :
D'autre part, en ce point la fonction doit être convexe, ce qui impose[10] :
troisième condition de criticité :
soit :
Les première et deuxième conditions induisent les paramètres critiques[10],[22] :
On a : ces deux fonctions sont miroir l'une de l'autre et se croisent en = 0,5 avec = 2. Elles présentent chacune un maximum, en = 2/3 et en = 1/3 ; ce maximum vaut = 9/4 = 2,25 (cf. figure 11)[10]. Tracés l'un en fonction de l'autre, les paramètres critiques délimitent dans la figure 12 le domaine dans lequel les paramètres de Margules permettent une démixtion et celui dans lequel ils ne le permettent pas[22].
La troisième condition est vraie quelle que soit pour le modèle de Margules. Selon ce modèle, il est donc possible de trouver des conditions critiques de démixtion pour toute composition de la phase liquide. Cette troisième contrainte réduit le domaine des compositions possibles d'apparition de la démixtion pour d'autres modèles d'enthalpie libre d'excès molaire. Pour certains modèles elle peut même ne jamais être vérifiée, ce qui rend ces modèles inaptes à représenter des démixtions[10].
Avec le modèle à un paramètre, , la deuxième condition implique que , soit = 0,5. La première condition donne = 2. Ce modèle ne génère qu'un seul point critique[26].
Figure 13 - Exemple de démixtion. Entre les points A et B, le mélange se sépare en deux phases. Avec = 1 et = 3, on a = 0,06 et = 0,66.
La figure 13 ci-contre montre un exemple de démixtion de phases. L'enthalpie libre de mélange molaire calculée avec le modèle de Margules (courbe bleue), à pression et température constantes, est concave sur une plage de composition située entre deux plages convexes. La fonction réelle doit rester dérivable et convexe sur l'ensemble des compositions. Pour cela, entre les points A et B elle ne suit pas la courbe bleue, mais le segment de droite violet. Aux points A et B le segment violet est tangent à la courbe bleue, les dérivées de l'enthalpie libre de mélange molaire doivent être égales à la pente du segment, d'où les deux équations permettant de déterminer et [11] :
Équilibre de deux phases démixées
soit :
Le modèle de Margules donne :
En amont de A il n'existe qu'une seule phase , dont la composition peut varier de 0 à . De même, en aval de B il n'existe qu'une seule phase , dont la composition peut varier de à 1. Pour et , l'enthalpie libre de mélange molaire vaut :
Entre A et B, au point P quelconque de composition , les deux phases , de composition , et , de composition , sont en équilibre. Avec une quantité de phase et une quantité de phase , la règle des moments donne la fraction de phase :
Pour , l'enthalpie libre de mélange molaire vaut :
Note - Les deux points A et B peuvent également être déterminés par les égalités[11],[27] :
Équilibre de deux phases démixées
avec et les coefficients d'activité des deux corps au point A, et et ceux au point B.
Figure 16 - Le point C est un hétéroazéotrope (équilibre liquide-liquide-vapeur), le point D un azéotrope négatif (équilibre liquide-vapeur).
Puisque le modèle de Margules permet de représenter des démixtions liquide-liquide, il permet la représentation d'équilibres liquide-liquide-vapeur[11]. La figure 14 montre les courbes de bulle et de rosée d'un mélange binaire telles qu'obtenues avec le modèle de Margules lorsque le modèle permet de déterminer une démixtion ; les paramètres et , et les points A et B sont les mêmes que ceux de la figure 13. Cette figure brute n'est pas réaliste dans sa partie supérieure et doit être modifiée[28].
Lorsque deux phases liquides sont en équilibre avec une troisième phase gazeuse, la règle des phases indique que, pour un mélange binaire, il n'y a qu'un seul degré de liberté. À température donnée, la pression et la composition d'équilibre ne peuvent donc être choisies, il s'agit d'un hétéroazéotrope[29]. Si la phase liquide a une composition et la phase liquide une composition , la pression et la composition de la vapeur au point d'hétéroazéotrope C sont déterminées selon[28] :
Pour des pressions supérieures à la pression ainsi déterminée, le mélange présente, selon la composition, un liquide de composition variable, un équilibre d'un liquide de composition et d'un liquide de composition , ou un liquide de composition variable. Pour des pressions inférieures à , le mélange présente, selon la composition, une phase liquide ou , un équilibre liquide-vapeur ou une phase gazeuse. Pour des pressions encore plus basses, le mélange ne présente qu'une unique phase gazeuse quelle que soit la composition. La figure 15 montre le diagramme de phases corrigé ; seules les courbes bleue et orange sont calculées avec le modèle de Margules, elles sont tronquées pour les pressions supérieures à . Les deux segments verticaux noirs tracés à partir des points A et B, qui délimitent les domaines d'existence des phases liquides et seules, et celui de leur coexistence, ne dépendent pas de la pression car le modèle de Margules ne dépend pas de la pression[28].
Il existe des systèmes (comme le système eau-butanone[30]) présentant, à température donnée, une démixtion ou un azéotrope en fonction de la composition et de la pression. La figure 16 montre un tel comportement calculé avec le modèle de Margules.
↑(en) Gilbert Newton Lewis et Merle Randall, Thermodynamics and the free energy of chemical substances, McGraw-Hill Book Company Inc., .
↑ abcde et f(en) Ronald W. Rousseau, Handbook of Separation Process Technology, John Wiley & Sons, , 1024 p. (ISBN9780471895589, lire en ligne), p. 24-30.
↑ abcdefgh et i(en) J. P. O'Connell et J. M. Haile, Thermodynamics : Fundamentals for Applications, Cambridge University Press, , 1re éd. (ISBN9781139443173, lire en ligne), p. 215-216.
↑Jean-Noël Jaubert et Romain Privat, Modèles thermodynamiques pour le génie des procédés, ISTE Group, coll. « Génie des procédés », , 196 p. (ISBN9781784057503, lire en ligne), p. 115.
↑(en) Pradeep Ahuja, Chemical Engineering Thermodynamics, PHI Learning Private Limited, , 720 p. (ISBN9788120336377, lire en ligne), p. 454-458.
↑(en) Hasan Orbey et Stanley I. Sandler, Modeling Vapor-Liquid Equilibria : Cubic Equations of State and Their Mixing Rules, Cambridge University Press, coll. « Cambridge Series in Chemical Engineering », , 1re éd., 207 p. (ISBN9780521620277, lire en ligne), p. 11-17.
↑(en) Emmerich Wilhelm, « The Art and Science of Solubility Measurements : What Do We Learn? », Netsu Sokutei, vol. 39, no 2, , p. 61-86 (lire en ligne [PDF]).
↑(en) H. C. Van Ness et M. M. Abbott, « Vapor‐liquid equilibrium. : Part VI. Standard state fugacities for supercritical components. », AIChE J., vol. 25, no 4, , p. 645-653 (lire en ligne).
↑ ab et cSabine Rode, Séparations thermiques en génie des procédés : Distillation, air humide, séchage, Éditions Ellipses, , 480 p. (ISBN9782340059221, lire en ligne), p. 37.
↑(en) Ulrich K. Deiters et Thomas Kraska, High-Pressure Fluid Phase Equilibria : Phenomenology and Computation, Amsterdam/Boston, Elsevier, coll. « Supercritical Fluid Science and Technology », , 342 p. (ISBN978-0-444-56347-7, ISSN2212-0505, lire en ligne), p. 31.
↑(en) Alexandre C. Dimian, Costin Sorin Bildea et Anton A. Kiss, Integrated Design and Simulation of Chemical Processes, Elsevier, , 886 p. (ISBN9780444627087, lire en ligne), p. 245-247.
↑(en) J. Gmehling, U. Onken, W. Arlt, P. Grenzheuser, U. Weidlich, B. Kolbe et J. Rarey, Chemistry Data Series, vol. I : Vapor-Liquid Equilibrium Data Collection, Dechema, 1991–2014 (lire en ligne).
↑(en) Robert H. Perry et Don W. Green, Perry's Chemical Engineers' Handbook, New York, McGraw-Hill, , 7e éd., 13:20 (ISBN0-07-115982-7).
↑(en) Amiya K. Jana, Chemical process modelling and computer simulation, PHI Learning Private Limited, , 2e éd., 376 p. (ISBN9788120344778, lire en ligne), p. 150-156.
Cette source donne les paramètres pour de nombreux couples de produits. Les coefficients d'activité y sont exprimés avec le logarithme décimal, . Les paramètres doivent être multipliés par 2,303 pour trouver les valeurs des coefficients exprimés avec le logarithme népérien.
(de) Max Margules, « Über die Zusammensetzung der gesättigten Dämpfe von Misschungen », Sitzungsberichte der Kaiserliche Akadamie der Wissenschaften Wien Mathematisch-Naturwissenschaftliche Klasse II, vol. 104, , p. 1243–1278 (lire en ligne).
(en) N.P. Komninos et E.D. Rogdakis, « Geometrical investigation and classification of three-suffix margules binary mixtures including single and double azeotropy », Fluid Phase Equilibria, vol. 494, , p. 212-227 (DOI10.1016/j.fluid.2019.04.017, lire en ligne).
(en) Biswajit Mukhopadhyay, Sabyasachi Basu et Michael J. Holdaway, « A discussion of Margules-type formulations for multicomponent solutions with a generalized approach », Geochimica et Cosmochimica Acta, vol. 57, , p. 227-283 (DOI10.1016/0016-7037(93)90430-5, lire en ligne).
Dans cet article, la convention prise est .
(en) Jaime Wisniak, « Liquid-liquid phase splitting - I : analytical models for critical mixing and azeotropy », Chemical Engineering Science, vol. 38, no 6, , p. 969–978 (DOI10.1016/0009-2509(83)80017-7, lire en ligne).
(en) Jean-Charles de Hemptinne et Jean-Marie Ledanois, Select Thermodynamic Models for Process Simulation : A Practical Guide Using a Three Steps Methodology, Éditions TECHNIP, coll. « IFP Énergies Nouvelles Publications - Institut français du pétrole publications - Publications (IFP Énergies nouvelles) », , 380 p. (lire en ligne), p. 160-170.
Michel Soustelle, Transformations entre phases, vol. 5, ISTE Group, coll. « Génie des procédés / Thermodynamique chimique approfondie », , 240 p. (ISBN9781784051235, lire en ligne).
Dans cet ouvrage, p. 143-144, une solution strictement régulière est définie par les coefficients d'activité et .





























































![{\displaystyle \ln \gamma _{i}=\left({\partial \over \partial n_{i}}\left[{n{\bar {G}}^{\text{E}} \over RT}\right]\right)_{P,T,n_{\neq i}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3cfc20be505b243d860e37d8d2ef11b2ca7fb3ee)
![{\displaystyle RT\,\ln \gamma _{i}=\left({\partial \left[n{\bar {G}}^{\text{E}}\right] \over \partial n_{i}}\right)_{P,T,n_{\neq i}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/4142d4c10727e1c05a6e75dbcd8d57266bfd2647)














![{\displaystyle {\bar {G}}_{i}^{\text{E}}=\left({\partial \left[n{\bar {G}}^{\text{E}}\right] \over \partial n_{i}}\right)_{P,T,n_{\neq i}}=RT\,\ln \gamma _{i}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7f89eb71e47d38e8cd9715d16f3ca22652758147)


























































































![Figure 9 - Enthalpie libre de mélange molaire avec le modèle d'enthalpie libre d'excès molaire de Margules à un paramètre (solution régulière). Sous la courbe critique, le mélange ne présente qu'une seule phase. Au-dessus de la courbe critique, le mélange présente une démixtion de deux phases[26].](//upload.wikimedia.org/wikipedia/commons/thumb/a/a1/Margules_-_d%C3%A9mixtion_1_param%C3%A8tre.jpg/574px-Margules_-_d%C3%A9mixtion_1_param%C3%A8tre.jpg)
 Figure 10 - Quelques courbes d'enthalpie libre de mélange molaire du modèle à deux paramètres de Margules. Pour = 2, les courbes avec = -0,6469 et = 2 sont des courbes de démixtion critiques présentant un point critique.
Figure 10 - Quelques courbes d'enthalpie libre de mélange molaire du modèle à deux paramètres de Margules. Pour = 2, les courbes avec = -0,6469 et = 2 sont des courbes de démixtion critiques présentant un point critique.![Figure 9 - Enthalpie libre de mélange molaire avec le modèle d'enthalpie libre d'excès molaire de Margules à un paramètre (solution régulière). Sous la courbe critique, le mélange ne présente qu'une seule phase. Au-dessus de la courbe critique, le mélange présente une démixtion de deux phases[26].](/wiki/Fichier:Margules_-_d%C3%A9mixtion_1_param%C3%A8tre.jpg)







![Figure 11 - Selon la composition[10].](//upload.wikimedia.org/wikipedia/commons/thumb/c/c6/Margules_-_param%C3%A8tres_critiques_de_d%C3%A9mixtion_-_1.jpg/574px-Margules_-_param%C3%A8tres_critiques_de_d%C3%A9mixtion_-_1.jpg)
![Figure 12 - Domaines de démixtion et d'impossibilité de démixtion[22].](//upload.wikimedia.org/wikipedia/commons/thumb/7/73/Margules_-_param%C3%A8tres_critiques_de_d%C3%A9mixtion_-_2.jpg/576px-Margules_-_param%C3%A8tres_critiques_de_d%C3%A9mixtion_-_2.jpg)
![Figure 11 - Selon la composition[10].](/wiki/Fichier:Margules_-_param%C3%A8tres_critiques_de_d%C3%A9mixtion_-_1.jpg)
![Figure 12 - Domaines de démixtion et d'impossibilité de démixtion[22].](/wiki/Fichier:Margules_-_param%C3%A8tres_critiques_de_d%C3%A9mixtion_-_2.jpg)









































 Figure 16 - Le point C est un hétéroazéotrope (équilibre liquide-liquide-vapeur), le point D un azéotrope négatif (équilibre liquide-vapeur).
Figure 16 - Le point C est un hétéroazéotrope (équilibre liquide-liquide-vapeur), le point D un azéotrope négatif (équilibre liquide-vapeur).











