
Secreted miR-153 Controls Proliferation and Invasion of Higher Gleason Score Prostate Cancer

 , , ,
, , ,  , and
, and 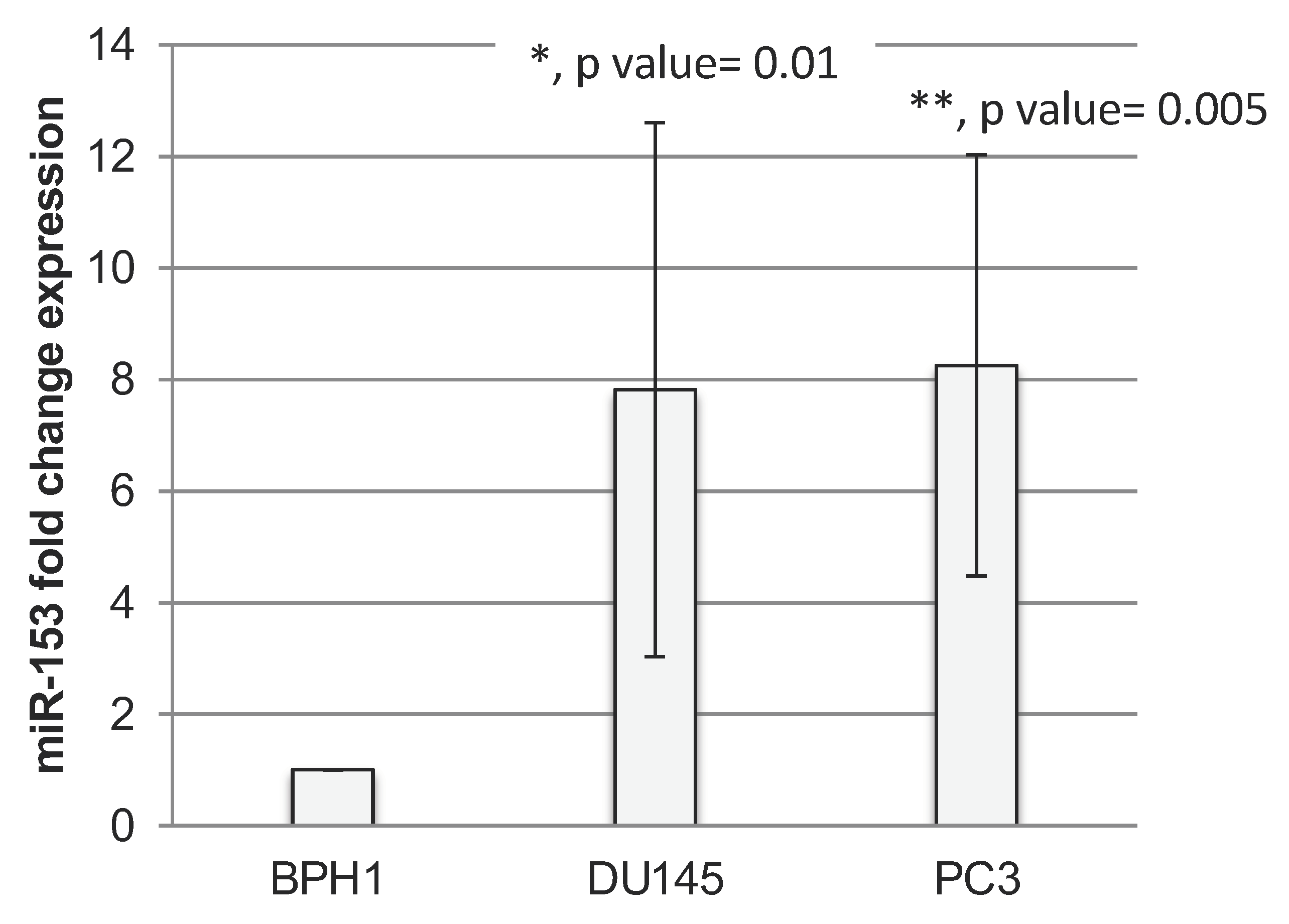{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
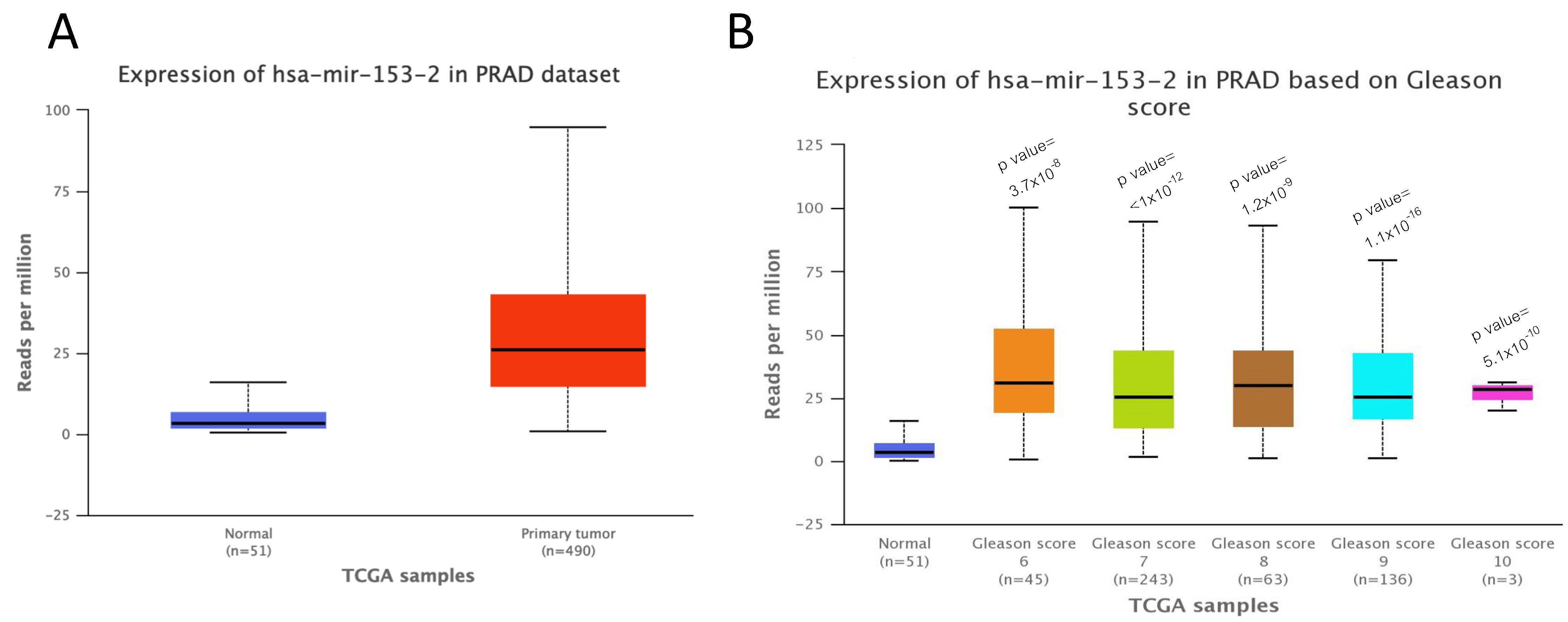
2.1. miR-153 Is Overexpressed in PC Cells with Higher Gleason Score
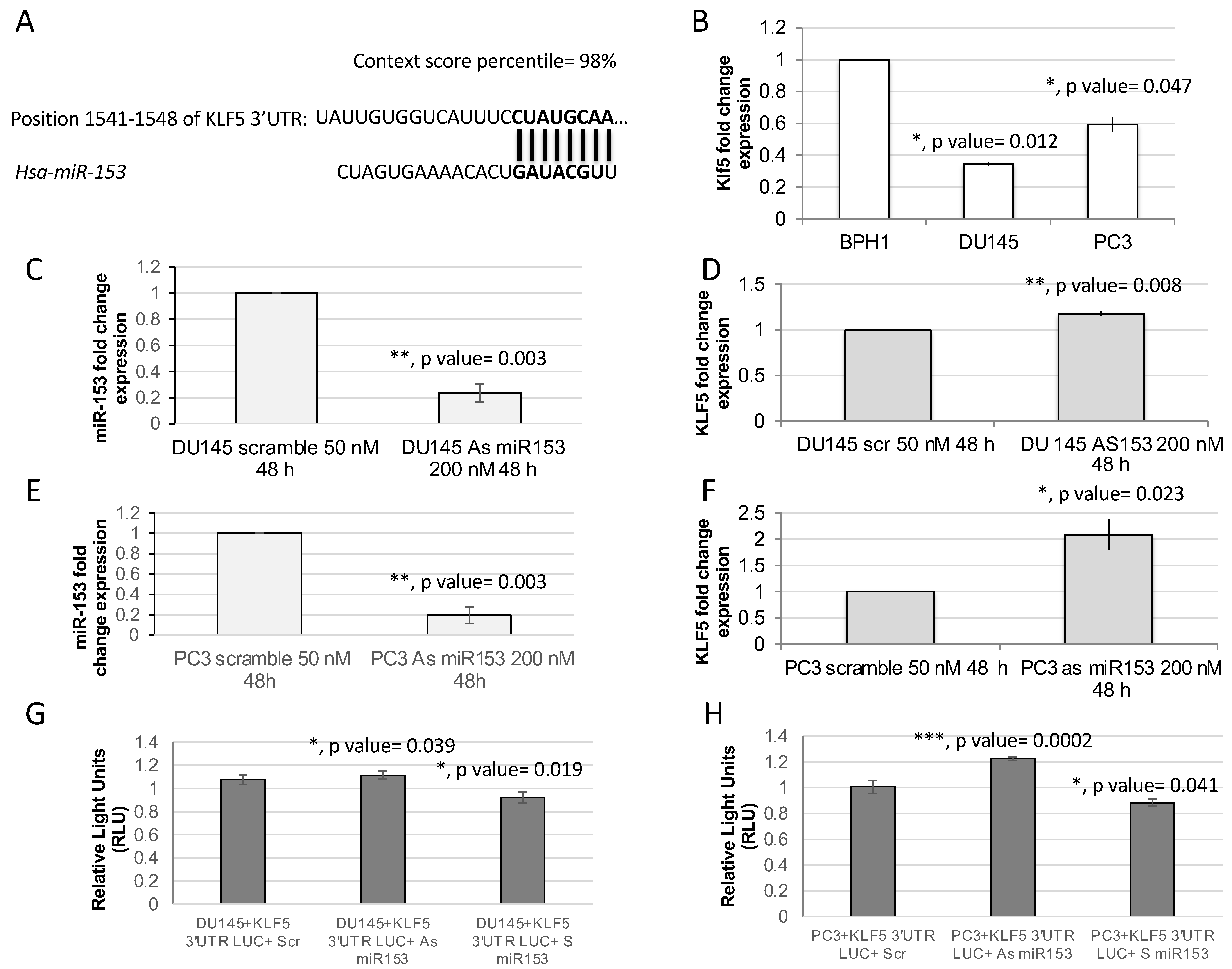
2.2. KLF5 Is a Direct Target of miR-153
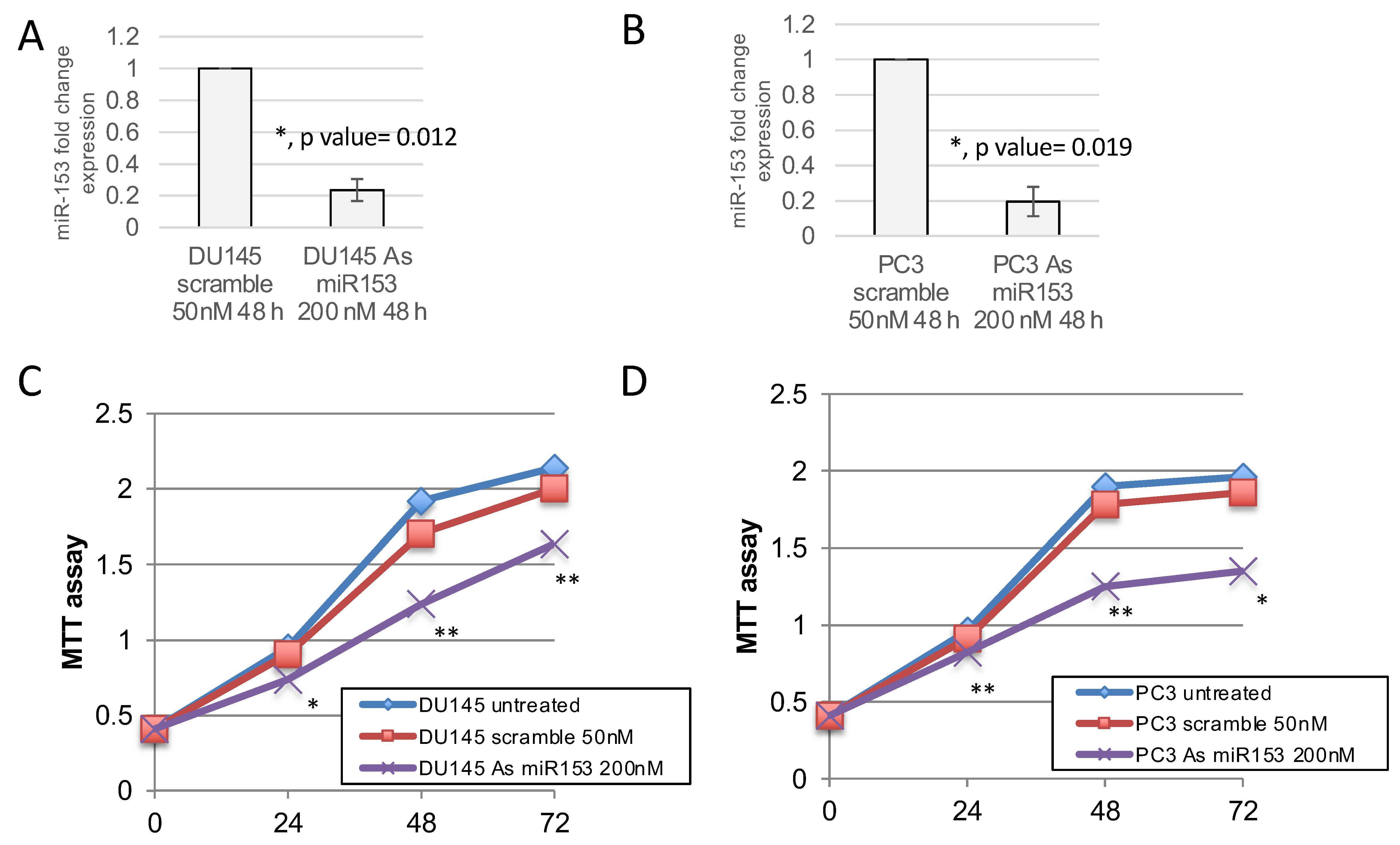
2.3. miR-153 Promotes Cell Proliferation in Higher GS Cells
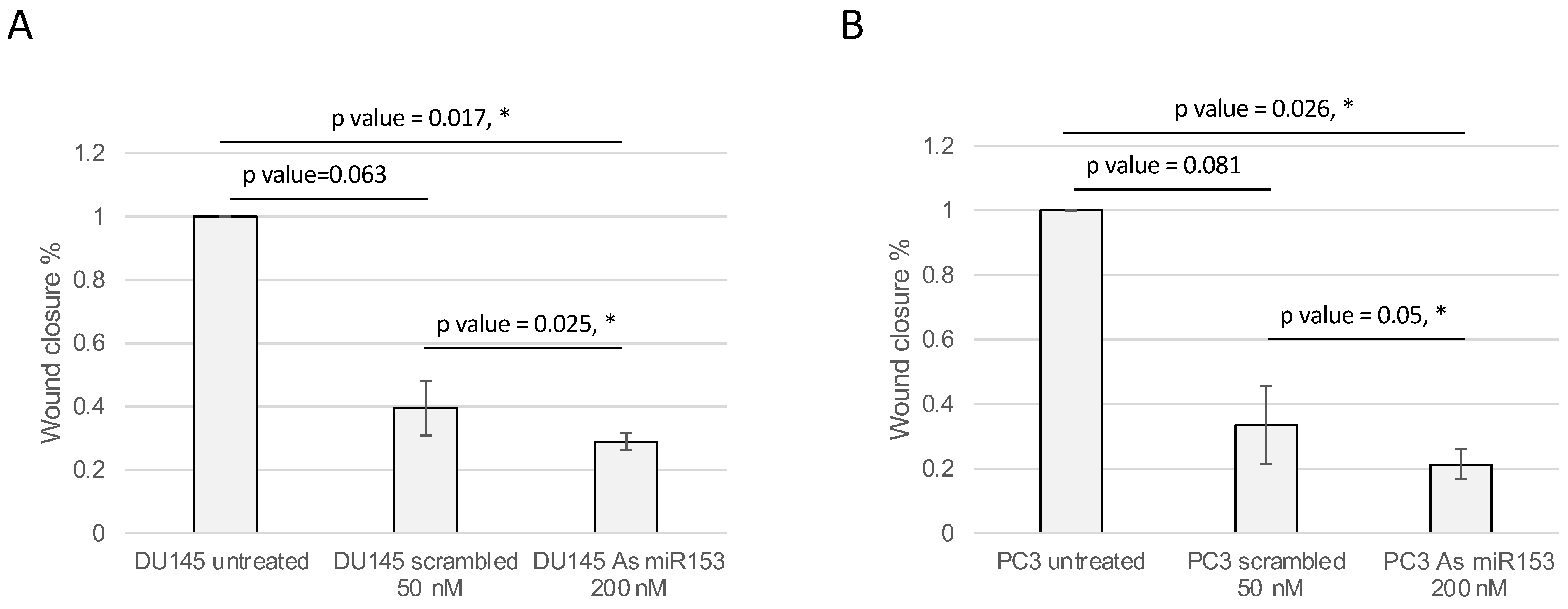
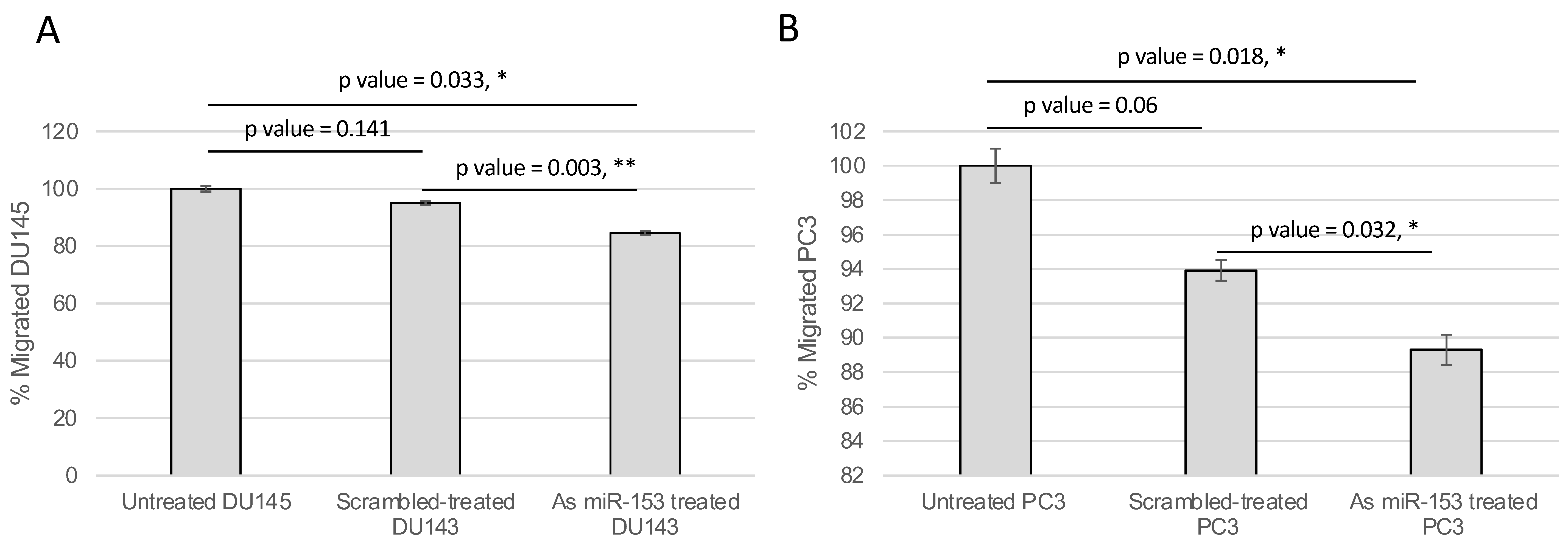
2.4. miR-153 Regulates Cell Migration and Invasion of PC Cells
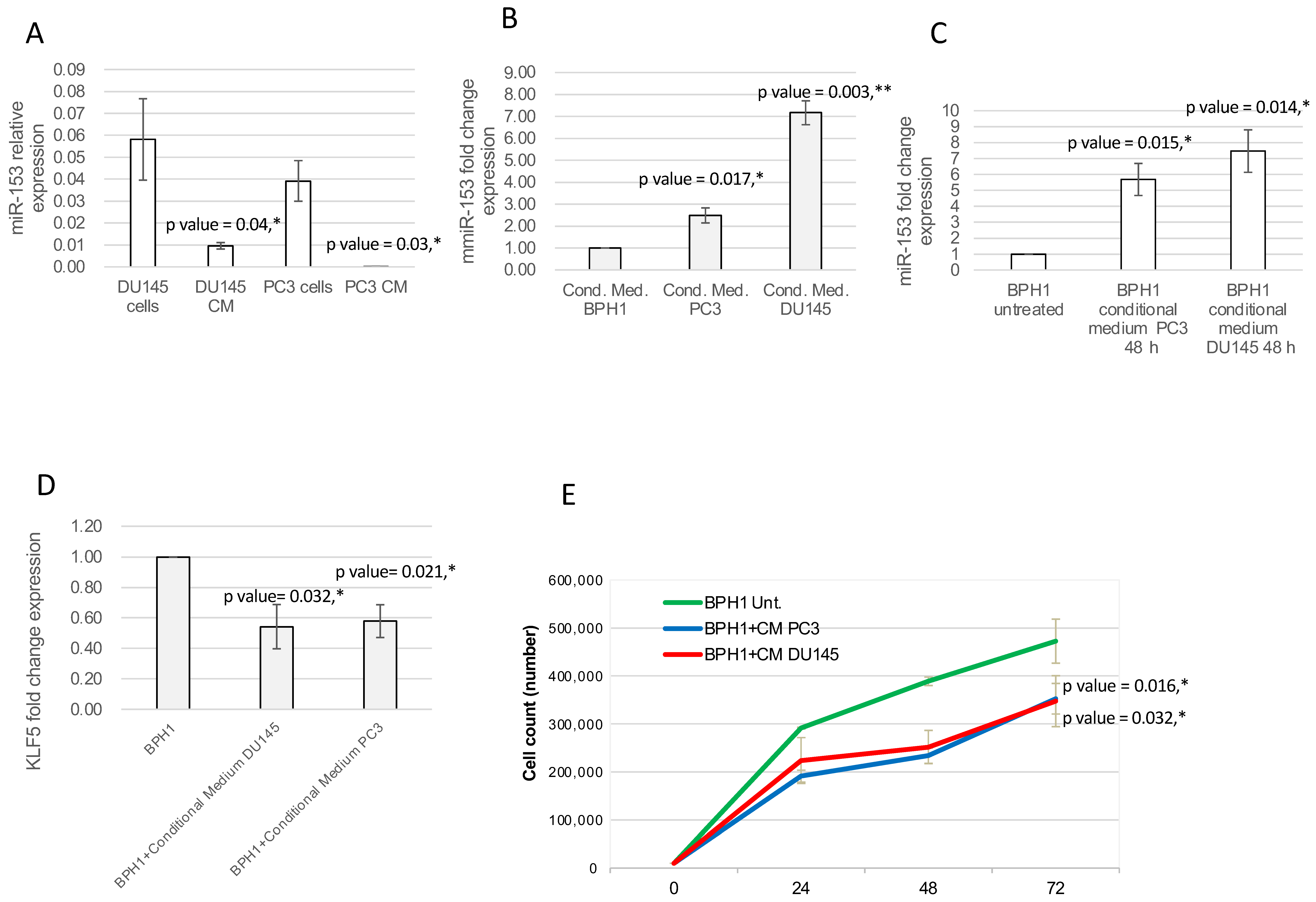
2.5. miR-153 Can Be Secreted, Influencing Surrounding Tissue
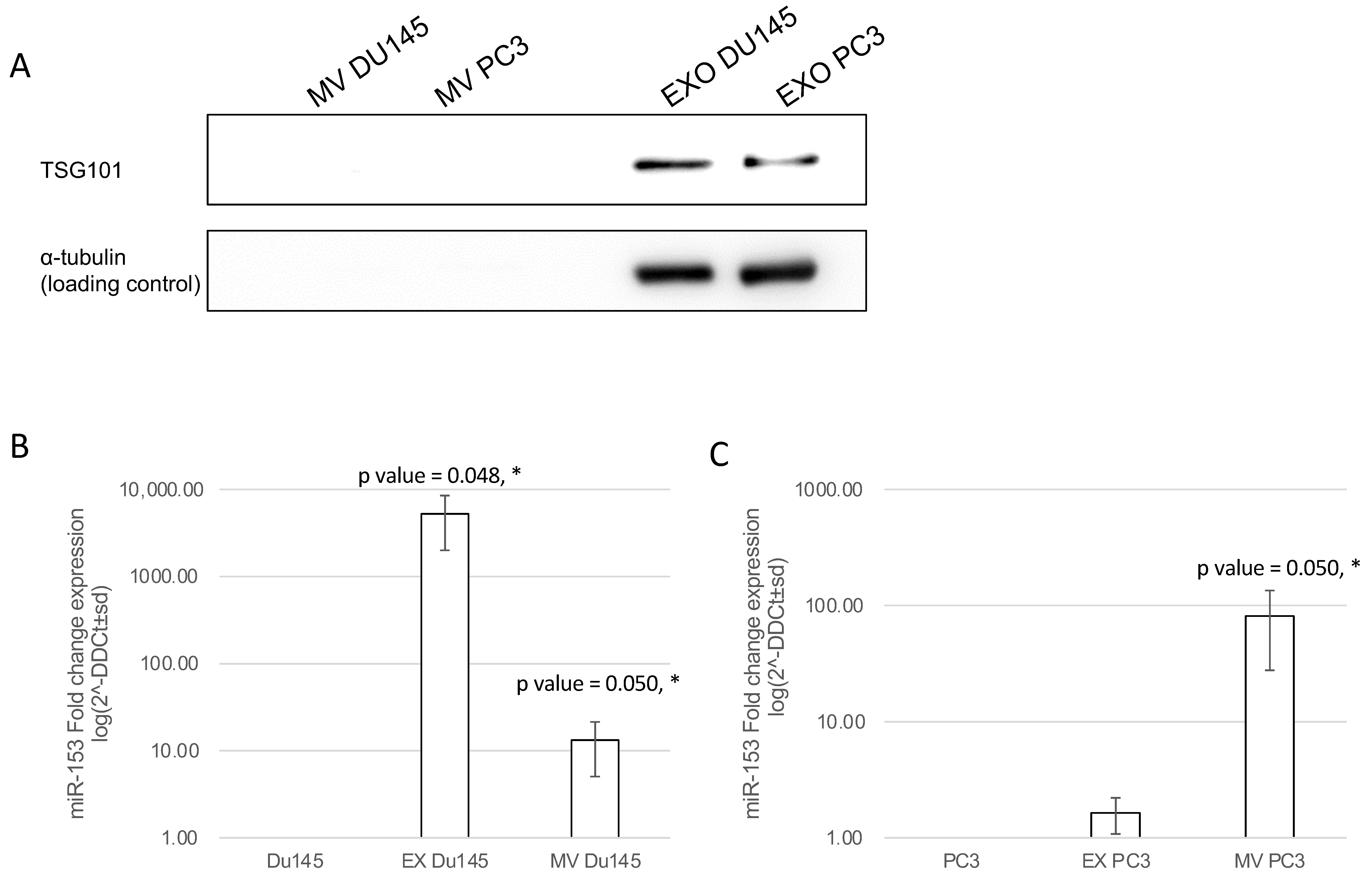
2.6. Exosomal and Microvesicle Distribution of Secreted miR-153
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Oligonucleotide Cell Treatment
4.3. UALCAN Analysis
4.4. Luciferase Reporter Assay
4.5. MTT Assays
4.6. Wound Healing and Boyden’s Chamber
4.7. Conditional Medium Treatment and Circulating miRNA Isolation
4.8. Cell Count
4.9. Exosomes (Ex) and Microvesicles (MV) Isolation by Differential Ultracentrifugation
4.10. SDS-PAGE and Western Blot
4.11. RNA Extraction and RT-qPCR Analysis
4.12. Analysis of miR-153 Secretion in Exosomes and Microvesicles
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stabile, A.; Giganti, F.; Rosenkrantz, A.B.; Taneja, S.S.; Villeirs, G.; Gill, I.S.; Allen, C.; Emberton, M.; Moore, C.M.; Kasivisvanathan, V. Multiparametric MRI for prostate cancer diagnosis: Current status and future directions. Nat. Rev. Urol. 2020, 17, 41–61. [Google Scholar] [CrossRef]
- Nepple, K.G.; Wahls, T.L.; Hillis, S.L.; Joudi, F.N. Gleason score and laterality concordance between prostate biopsy and prostatectomy specimens. Int. Braz. J. Urol. 2009, 35, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Damascelli, A.; Gallivanone, F.; Cristel, G.; Cava, C.; Interlenghi, M.; Esposito, A.; Brembilla, G.; Briganti, A.; Montorsi, F.; Castiglioni, I.; et al. Advanced Imaging Analysis in Prostate MRI: Building a Radiomic Signature to Predict Tumor Aggressiveness. Diagnostics 2021, 11, 594. [Google Scholar] [CrossRef]
- Van Poppel, H.; Roobol, M.J.; Chapple, C.R.; Catto, J.W.F.; N’Dow, J.; Sonksen, J.; Stenzl, A.; Wirth, M. Prostate-specific Antigen Testing as Part of a Risk-Adapted Early Detection Strategy for Prostate Cancer: European Association of Urology Position and Recommendations for 2021. Eur. Urol. 2021, 80, 703–711. [Google Scholar] [CrossRef]
- Vanacore, D.; Boccellino, M.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Di Franco, R.; Romano, F.J.; Montanari, M.; La Mantia, E.; Piscitelli, R.; et al. Micrornas in prostate cancer: An overview. Oncotarget 2017, 8, 50240–50251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramovic, I.; Ulamec, M.; Katusic Bojanac, A.; Bulic-Jakus, F.; Jezek, D.; Sincic, N. miRNA in prostate cancer: Challenges toward translation. Epigenomics 2020, 12, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.A.; Valera, V.A.; Pinto, P.A.; Merino, M.J. Comprehensive microRNA Profiling of Prostate Cancer. J. Cancer 2013, 4, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Bertoli, G.; Cava, C.; Castiglioni, I. MicroRNAs as Biomarkers for Diagnosis, Prognosis and Theranostics in Prostate Cancer. Int. J. Mol. Sci. 2016, 17, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Li, R.S.; Li, Y.H.; Zhong, S.; Chen, Y.Y.; Zhang, C.M.; Hu, M.M.; Shen, Z.J. miR-21 as an independent biochemical recurrence predictor and potential therapeutic target for prostate cancer. J. Urol. 2012, 187, 1466–1472. [Google Scholar] [CrossRef] [PubMed]
- Bidarra, D.; Constancio, V.; Barros-Silva, D.; Ramalho-Carvalho, J.; Moreira-Barbosa, C.; Antunes, L.; Mauricio, J.; Oliveira, J.; Henrique, R.; Jeronimo, C. Circulating MicroRNAs as Biomarkers for Prostate Cancer Detection and Metastasis Development Prediction. Front. Oncol. 2019, 9, 900. [Google Scholar] [CrossRef] [Green Version]
- Stoen, M.J.; Andersen, S.; Rakaee, M.; Pedersen, M.I.; Ingebriktsen, L.M.; Bremnes, R.M.; Donnem, T.; Lombardi, A.P.G.; Kilvaer, T.K.; Busund, L.T.; et al. High expression of miR-17-5p in tumor epithelium is a predictor for poor prognosis for prostate cancer patients. Sci. Rep. 2021, 11, 13864. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.W.; Zhang, G.Y.; Bai, Y.; Zhao, B.; Yang, H. Increased expression of miR-153 predicts poor prognosis for patients with prostate cancer. Medicine 2019, 98, e16705. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Cava, C.; Bertoli, G.; Colaprico, A.; Bontempi, G.; Mauri, G.; Castiglioni, I. In-Silico Integration Approach to Identify a Key miRNA Regulating a Gene Network in Aggressive Prostate Cancer. Int. J. Mol. Sci. 2018, 19, 910. [Google Scholar] [CrossRef] [Green Version]
- Tarapore, R.S.; Yang, Y.; Katz, J.P. Restoring KLF5 in esophageal squamous cell cancer cells activates the JNK pathway leading to apoptosis and reduced cell survival. Neoplasia 2013, 15, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Chang, L.Y.; Zhao, S.; Zhao, J.J.; Xiong, Y.J.; Cao, F.Y.; Yuan, L.; Zhang, Q.; Wang, X.Y.; Geng, M.L.; et al. KLF5 promotes cervical cancer proliferation, migration and invasion in a manner partly dependent on TNFRSF11a expression. Sci. Rep. 2017, 7, 15683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; He, B.; He, J.; Mao, X. Upregulation of miR-153 promotes cell proliferation via downregulation of the PTEN tumor suppressor gene in human prostate cancer. Prostate 2013, 73, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Chen, C. The roles and regulation of the KLF5 transcription factor in cancers. Cancer Sci. 2021, 112, 2097–2117. [Google Scholar] [CrossRef] [PubMed]
- Pattison, J.M.; Posternak, V.; Cole, M.D. Transcription Factor KLF5 Binds a Cyclin E1 Polymorphic Intronic Enhancer to Confer Increased Bladder Cancer Risk. Mol. Cancer Res. 2016, 14, 1078–1086. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, B.; Liu, M.; Fu, X.; Ci, X.; A, J.; Fu, C.; Dong, G.; Wu, R.; Zhang, Z.; et al. KLF5 Is Crucial for Androgen-AR Signaling to Transactivate Genes and Promote Cell Proliferation in Prostate Cancer Cells. Cancers 2020, 12, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liang, S.; Duan, X. Molecular mechanism of miR-153 inhibiting migration, invasion and epithelial-mesenchymal transition of breast cancer by regulating transforming growth factor beta (TGF-beta) signaling pathway. J. Cell Biochem. 2019, 120, 9539–9546. [Google Scholar] [CrossRef]
- Xia, W.; Ma, X.; Li, X.; Dong, H.; Yi, J.; Zeng, W.; Yang, Z. miR-153 inhibits epithelial-to-mesenchymal transition in hepatocellular carcinoma by targeting Snail. Oncol. Rep. 2015, 34, 655–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaprico, A.; Olsen, C.; Bailey, M.H.; Odom, G.J.; Terkelsen, T.; Silva, T.C.; Olsen, A.V.; Cantini, L.; Zinovyev, A.; Barillot, E.; et al. Interpreting pathways to discover cancer driver genes with Moonlight. Nat. Commun. 2020, 11, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Pickard, K.; Jenei, V.; Bullock, M.D.; Bruce, A.; Mitter, R.; Kelly, G.; Paraskeva, C.; Strefford, J.; Primrose, J.; et al. miR-153 supports colorectal cancer progression via pleiotropic effects that enhance invasion and chemotherapeutic resistance. Cancer Res. 2013, 73, 6435–6447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, Y.; Yuan, W.; Qiu, S. MicroRNA-153 functions as a tumor suppressor in gastric cancer via targeting Kruppel-like factor 5. Exp. Ther. Med. 2022, 23, 266. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Shin, H.; Moon, H.W.; Park, Y.H.; Park, J.; Lee, J.Y. Urinary exosomal microRNA profiling in intermediate-risk prostate cancer. Sci. Rep. 2021, 11, 7355. [Google Scholar] [CrossRef] [PubMed]
- Barcelo, M.; Castells, M.; Bassas, L.; Vigues, F.; Larriba, S. Semen miRNAs Contained in Exosomes as Non-Invasive Biomarkers for Prostate Cancer Diagnosis. Sci. Rep. 2019, 9, 13772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, N.H.; Abdellateif, M.S.; Thabet, G.; Kassem, S.H.; El-Salam, M.A.; El-Leithy, A.A.; Selim, M.M. Combining PHI and miRNAs as Biomarkers in Prostate Cancer Diagnosis and Prognosis. Clin. Lab. 2019, 65, 1159–1168. [Google Scholar] [CrossRef]
- Gordetsky, J.; Epstein, J. Grading of prostatic adenocarcinoma: Current state and prognostic implications. Diagn. Pathol. 2016, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grada, A.; Otero-Vinas, M.; Prieto-Castrillo, F.; Obagi, Z.; Falanga, V. Research Techniques Made Simple: Analysis of Collective Cell Migration Using the Wound Healing Assay. J. Investig. Dermatol. 2017, 137, e11–e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoli, G.; Cava, C.; Corsi, F.; Piccotti, F.; Martelli, C.; Ottobrini, L.; Vaira, V.; Castiglioni, I. Triple negative aggressive phenotype controlled by miR-135b and miR-365: New theranostics candidates. Sci. Rep. 2021, 11, 6553. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertoli, G.; Panio, A.; Cava, C.; Gallivanone, F.; Alini, M.; Strano, G.; Molfino, F.; Brioschi, L.; Viani, P.; Porro, D. Secreted miR-153 Controls Proliferation and Invasion of Higher Gleason Score Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 6339. https://doi.org/10.3390/ijms23116339
Bertoli G, Panio A, Cava C, Gallivanone F, Alini M, Strano G, Molfino F, Brioschi L, Viani P, Porro D. Secreted miR-153 Controls Proliferation and Invasion of Higher Gleason Score Prostate Cancer. International Journal of Molecular Sciences. 2022; 23(11):6339. https://doi.org/10.3390/ijms23116339
Chicago/Turabian StyleBertoli, Gloria, Antonella Panio, Claudia Cava, Francesca Gallivanone, Martina Alini, Giulia Strano, Federico Molfino, Loredana Brioschi, Paola Viani, and Danilo Porro. 2022. "Secreted miR-153 Controls Proliferation and Invasion of Higher Gleason Score Prostate Cancer" International Journal of Molecular Sciences 23, no. 11: 6339. https://doi.org/10.3390/ijms23116339